Changing Therapeutic Landscape of HAE due to C1 inhibitor Deficiency - Twitter summary from the 2019 WSAAI meeting
Bruce Zuraw presented on "The Ever-Changing Therapeutic Landscape of HAE due to C1 inhibitor Deficiency.”
C1-inhibitor treatment has been exploding in recent years. And for a good reason: C1-inhibitor replacement works better than androgen therapy for HAE, and is safer.
3 treatment strategies - on demand, ST prophylaxis and LT prophylaxis. Traditionally, there have been three main treatment strategies for HAE-C1INH: https://twitter.com/MatthewBowdish/status/1088611625723023360
Summary of treatment landscape up until 2 years ago: https://twitter.com/RayFirszt/status/1088612218776633344
New meds for HAE - Haegarda and lanadelumab hold promise that HAE patients can live a pretty much normal life.
HAEgarda and lanadelumab appear to be more effective than Cinryze and are cheaper.
Haegarda is a SC C1-inhibitor - 60 IU/kg - it reduced attacks by 95% . 80% of patients were attack free. Safe. Haegarda was associated with injection site reactions. Dosing is 2 times per week.
Functional C1-inhibitor levels correlate with risk of attack. We should measure the C1-INH function in patients who are treated and are not well controlled.
Lanadelumab dose is 300 mg SC q2 weeks. Kallikrein inhibitor. Significant reduction in attacks. Also attacks were less severe.
Upcoming meds for HAE: oral kallikrein inhibitor in a phase 2 study. Seems to be effective (BCX7353). Dose is 125 mg taken orally. Higher doses seem to be irritating to GI tract. Will be used for LTP. BCX7353 in Phase II Trial, oral plasma kallikrein inhibitor, results in NEJM: https://twitter.com/MatthewBowdish/status/1088618463931785216
Ruconest is being looked at for LTP for HAE. Dosing twice per week showed a 60-70% reduction of attacks in severe patients. 1 dose per week led to a 40% reduction - lower effectiveness likely to short half life.
CSL312, a monoclonal Factor XII inhibitor, is in early stages of Phase II trial. It inhibits bradykinin formation through contact system.
Another possible future option is an Antisense Oligonucleotide (ASO) against Prekallikrein, currently in Phase I: https://twitter.com/MatthewBowdish/status/1088621097661038592
Direct medical costs of on demand tx of HAE plus indirect costs equal roughly $400K/year, which rivals those expensive preventative meds.
Is gene editing an answer to HAE? Gene therapy was being studied in C1-inhibitor def. Adenovirus-associated gene replacement. Currently on HOLD.
HAE is a pediatric disease: average age of symptom onset is 10-11 years. Some are very young. The younger the age, possibly more severe dz. No drugs approved for HAE under 12: https://twitter.com/RayFirszt/status/1088614626764283904
This is a Twitter summary from the 2019 WSAAI meeting. This summary was compiled from the tweets posted by Matthew Bowdish @MatthewBowdish and Ray Firszt @RayFirszt, who attended the 2019 Western Society of Allergy, Asthma and Immunology (WSAAI) meeting. The tweets were labeled #WSAAI. The text was edited by me.
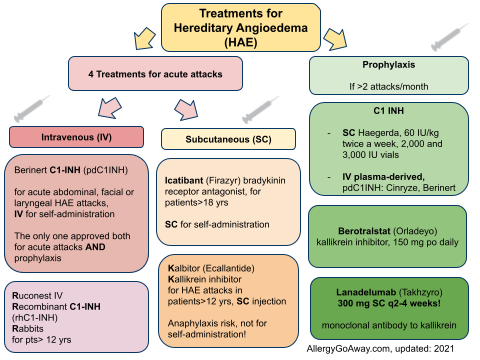
Treatments for Hereditary Angioedema (HAE) (click to enlarge the image).
C1-inhibitor treatment has been exploding in recent years. And for a good reason: C1-inhibitor replacement works better than androgen therapy for HAE, and is safer.
3 treatment strategies - on demand, ST prophylaxis and LT prophylaxis. Traditionally, there have been three main treatment strategies for HAE-C1INH: https://twitter.com/MatthewBowdish/status/1088611625723023360
Summary of treatment landscape up until 2 years ago: https://twitter.com/RayFirszt/status/1088612218776633344
New meds for HAE - Haegarda and lanadelumab hold promise that HAE patients can live a pretty much normal life.
HAEgarda and lanadelumab appear to be more effective than Cinryze and are cheaper.
Haegarda is a SC C1-inhibitor - 60 IU/kg - it reduced attacks by 95% . 80% of patients were attack free. Safe. Haegarda was associated with injection site reactions. Dosing is 2 times per week.
Functional C1-inhibitor levels correlate with risk of attack. We should measure the C1-INH function in patients who are treated and are not well controlled.
Lanadelumab dose is 300 mg SC q2 weeks. Kallikrein inhibitor. Significant reduction in attacks. Also attacks were less severe.
Upcoming meds for HAE: oral kallikrein inhibitor in a phase 2 study. Seems to be effective (BCX7353). Dose is 125 mg taken orally. Higher doses seem to be irritating to GI tract. Will be used for LTP. BCX7353 in Phase II Trial, oral plasma kallikrein inhibitor, results in NEJM: https://twitter.com/MatthewBowdish/status/1088618463931785216
Ruconest is being looked at for LTP for HAE. Dosing twice per week showed a 60-70% reduction of attacks in severe patients. 1 dose per week led to a 40% reduction - lower effectiveness likely to short half life.
CSL312, a monoclonal Factor XII inhibitor, is in early stages of Phase II trial. It inhibits bradykinin formation through contact system.
Another possible future option is an Antisense Oligonucleotide (ASO) against Prekallikrein, currently in Phase I: https://twitter.com/MatthewBowdish/status/1088621097661038592
Direct medical costs of on demand tx of HAE plus indirect costs equal roughly $400K/year, which rivals those expensive preventative meds.
Is gene editing an answer to HAE? Gene therapy was being studied in C1-inhibitor def. Adenovirus-associated gene replacement. Currently on HOLD.
HAE is a pediatric disease: average age of symptom onset is 10-11 years. Some are very young. The younger the age, possibly more severe dz. No drugs approved for HAE under 12: https://twitter.com/RayFirszt/status/1088614626764283904
This is a Twitter summary from the 2019 WSAAI meeting. This summary was compiled from the tweets posted by Matthew Bowdish @MatthewBowdish and Ray Firszt @RayFirszt, who attended the 2019 Western Society of Allergy, Asthma and Immunology (WSAAI) meeting. The tweets were labeled #WSAAI. The text was edited by me.
Treatments for Hereditary Angioedema (HAE) (click to enlarge the image).